
Chemistry and Materials Earth Sciences and Environment Engineering and Energy Fundamental physics Mathematics and Computer Science Medicine and Life Sciences All 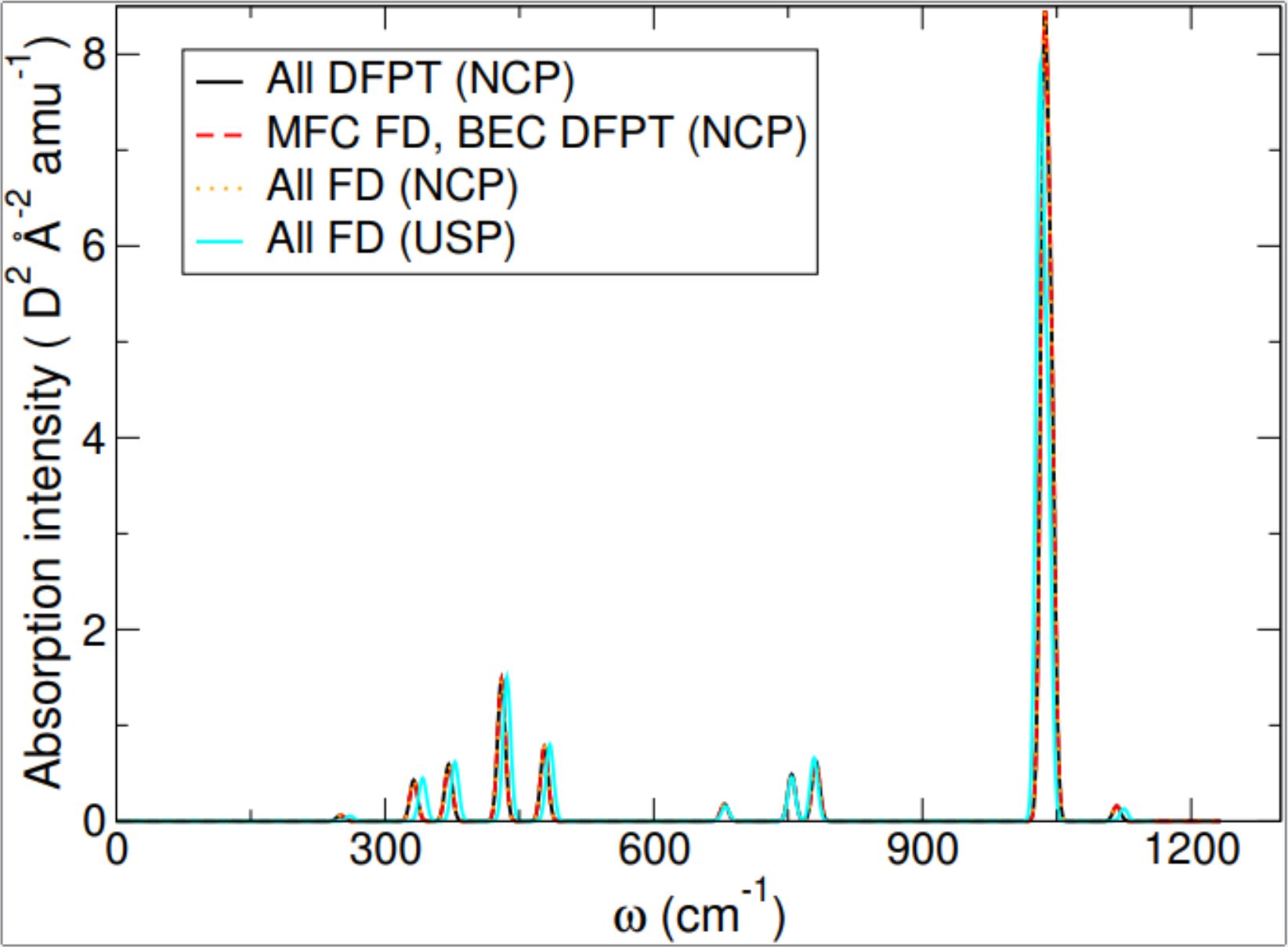
Computing infra-red spectra using finite differencing in CASTEP
ARCHER2-eCSE09-03 : Prof Jonathan R Yates (University of Oxford)Subject Area:
Chemistry and Materials Infra-red (IR) spectroscopy is used to gain insight into the structure of materials. By shining IR light on a material and mapping out how strongly the light is absorbed for different frequencies, it is possible to build up an IR spectrum which is characteristic of that material. However, it is difficult to work backwards from the experimental spectrum data to determine the structure of a given material. Instead, first principles materials modelling methods can be used to simulate computationally what the IR spectrum would look like for a proposed structure, and this can then be compared against experimental data. This eCSE project added new functionality to CASTEP, a leading code for calculating the properties of materials from first principles. The new functionality allows IR spectra to be simulated for materials for which this was not previously possible. This will be of immediate benefit to researchers in both academia and industry who use IR spectroscopy, opening up new opportunities to compare computational spectra with experimental ones. Read more...

Improving multi-threaded scaling of CONQUEST
ARCHER2-eCSE08-10 : Prof David R Bowler (University College London)Subject Area:
Chemistry and Materials Density functional theory (DFT) is a widely used technique for the modelling of materials. It can be used to predict the atomic and electronic structure of materials in areas as diverse as physics, chemistry, earth sciences, and biochemistry. Standard computational approaches to DFT can only be used to model systems of a few hundred atoms, which restricts the type of problem that can be studied. However, the CONQUEST DFT code is designed to model problems with very large numbers of atoms – from several thousand atoms to millions of atoms. This eCSE project was designed to improve the performance of CONQUEST, so that it runs faster and on larger computers, enabling more challenging and interesting problems to be studied. The code is already massively parallel, but this work improves its performance on multi-core computers. Read more...
 slab (yellow spheres) with adsorbed CO molecule (black and red spheres)")
Improvements in periodic representation of solvated systems with FHI-aims
ARCHER2-eCSE08-03 : Dr Andrew Logsdail (Cardiff University)Subject Area:
Chemistry and Materials Computational modelling can provide invaluable insights into chemical processes at the atomic scale. These insights can help researchers make new discoveries, for example in the search for new sustainable materials or new adsorbents for medicine. Many of the relevant chemical processes occur in liquids close to a solid surface. However, the complexity of the molecular interactions means that atomistic modelling of liquids is a notoriously difficult task, and computational models must balance accuracy with computational cost. FHI-aims is a widely adopted code for quantum chemistry and materials science, and is especially efficient for simulation of solid surfaces. This project enhanced the capability of FHI-aims to simulate solid surfaces within liquids, improving usability and performance, and allowing the closer alignment of computer models with real-life systems. Read more...

Improving the Performance of Linear-Scaling BigDFT for ARCHER2 and Beyond
ARCHER2-eCSE07-02 : Dr Laura E Ratcliff (University of Bristol)Subject Area:
Chemistry and Materials BigDFT is a free Density Functional Theory (DFT) software package which can be used to calculate the total energy, charge density and electronic structure of systems made of electrons and nuclei. It is designed for use on massively parallel computer architectures. BigDFT can provide insights into a diverse range of problems, from improving our understanding of materials for technological applications, to addressing challenges in biology, such as enzyme-based detoxification and comparing different SARS-CoV-2 mutations. LS-BigDFT, the linear scaling version of the code, enables first principles simulations of thousands of atoms, making it capable of simulating systems which are beyond the reach of most DFT codes. This eCSE project worked to address issues affecting the stability and performance of BigDFT on ARCHER2. The work has led to significant improvements in the code, enabling calculations to be performed for a range of system sizes and node counts which would not previously have been possible. The work has also led to significantly improved stability of BigDFT on ARCHER2. Read more...

Scaling up and coupling adaptive kinetic Monte Carlo with large-scale DFT
ARCHER2-eCSE05-04 : Prof Chris-Kriton Skylaris (University of Southampton)Subject Area:
Chemistry and Materials Diffusion of atoms and ions in solid state materials is essential for the function of products such as batteries, fuel cells and magnetic materials. Understanding atomic and/or ionic diffusion is therefore essential for the design and controlled manufacture of novel materials. However, computational techniques to study and analyse solid state diffusion are limited. The adaptive kinetic Monte Carlo method (aKMC) based on empirical potentials allows for material properties to be calculated efficiently, but it has clear limitations in its accuracy for certain chemical processes. In contrast, Density Functional Theory (DFT) calculations overcome these limitations, but are computationally more expensive. The recent availability of large numbers of cores on ARCHER2 provides an opportunity to combine these two approaches and develop methodologies capable of harnessing in excess of 100,000 cores. This eCSE project developed an interface to combine an aKMC program (ACDC) and a linear-scaling DFT program (ONETEP), making it possible to undertake large-scale aKMC simulations with a higher degree of accuracy than was previously possible. Read more...

Relativistic all-electron orbital-constrained Density Functional Theory to simulate x-ray photoemission and absorption spectroscopy
ARCHER2-eCSE04-03 : Prof Reinhard Maurer (University of Warwick)Subject Area:
Chemistry and Materials X-ray photoemission spectroscopy (XPS) is one of the most important materials characterization techniques that are used in fundamental research and industrial quality control. FHI-aims is a software package for computational molecular and materials science that provides advanced XPS simulation capabilities. It is used to simulate the chemical and physical properties of atoms, molecules, nanostructures, solids, and surfaces. Such simulations allow researchers to discover new materials, which may be stronger, cheaper or more durable than existing materials. FHI-aims can run efficiently on anything from a laptop to a supercomputer with tens of thousands of cores. However, the section of the code used for carrying out core-hole calculations was outdated and inefficient. The work done in this eCSE project has brought that part of the code up to date, so that it is now more efficient, better documented, and easier to maintain. These changes will significantly enhance the simulation of core-level spectroscopy, enabling the simulating of bigger and more detailed systems in future. The changes will also allow a much broader range of researchers to work on these types of simulations, thus ultimately enabling new scientific discoveries. Read more...
 new material TaFeSb. This is published in:<br> G. Naydenov et al., J. Phys. Mater. 2, 035002 (2019)<br>doi: 10.1088/2515-7639/ab16fb")
Future-proof parallelism in CASTEP
ARCHER2-eCSE03-15 : Dr Phil Hasnip (University of York)Subject Area:
Chemistry and Materials CASTEP is a leading materials modelling program, capable of predicting the electronic, mechanical and chemical properties of matter. The CASTEP code is widely used on UK High Performance Computing (HPC) facilities to tackle many scientific problems. One example of particular scientific interest in this area is the design of new thermoelectric materials. An efficient thermoelectric material could dramatically improve the energy efficiency of everyday appliances by using the Seebeck effect to convert waste heat into electrical power, but understanding and optimising thermoelectric performance requires computationally demanding simulations of the vibrational properties of the material. This eCSE project aimed to re-engineer the CASTEP code in order to substantially improve its performance on parallel computing systems, allowing calculations to be performed on ARCHER2 which were not previously possible. The improved version was tested on an example thermoelectric material simulation, which it completed in half the time of the original code. CASTEP is used by many industrial research sites, including household names such as Honda and Panasonic, as well as academic research groups. All the software developed during this project has been integrated into the main CASTEP code and released to the userbase worldwide. The work in this project will thus benefit all parallel CASTEP simulations, in both academic and industrial research settings. Read more...

Realising a modular data interface to couple quantum mechanical calculators with external data-driven workflows
ARCHER2-eCSE03-10 : Dr Andrew Logsdail (Cardiff University)Subject Area:
Chemistry and Materials Electronic structure calculations are the workhorse of modern computational chemistry, allowing for the prediction of information for a range of vital societal applications, such as drug efficacy, composition of new solar cells, or environmental impact of pollutants. However, the intrinsically complicated nature of electron interactions leads to a great variety of computational chemistry software. Complex interoperation between computer codes can slow the progress of research. To address this, a new applications programming interface, Atomic Simulation Interface (ASI), was developed and then implemented in established electronic structure software packages. This offers a unified and efficient way to export and import large data structures used in electronic structure calculations and for classical molecular dynamics simulations. Read more...
Unleashing the Potential for Superior Parallel Performance of the ONETEP Linear-Scaling Density Functional Theory Package on ARCHER2
ARCHER2-eCSE03-05 : Prof Nicholas D M Hine (University of Warwick)Subject Area:
Chemistry and Materials ONETEP is an ab initio electronic structure package which is widely used in academia and industry for large-scale atomistic simulations of systems such as nanomaterials, crystalline interfaces, and biological materials. Such simulations can be used to develop new drugs, or investigate properties of novel materials such as those used in batteries and photovoltaic cells. This eCSE project aimed to significantly improve the parallel scaling of the ONETEP Theory code when run on state-of-the-art parallel computing environments such as ARCHER2. Although the parallel performance gains of the implemented approaches were not as high as had been hoped, the project also produced other useful outputs, including documentation of best practice when running at very large scale with hybrid MPI/OpenMP parallelism. The work also fed successfully into further developments undertaken as part of the Software for Research Communities EPSRC project. While refocusing parallel scaling efforts, including new developments targeted at porting to GPUs, this new work has made great progress in improving the overall speed to the extent originally envisaged in the eCSE project. Read more...

Support for advanced transition state search techniques in CASTEP
ARCHER2-eCSE02-04 : Dr James R Kermode (University of Warwick)Subject Area:
Chemistry and Materials The CASTEP density functional theory code is a UK flagship code, specialised for solid materials, and is heavily used on ARCHER2 (300-400 active users). While support for obtaining the ground-state electronic and atomic configurations is now very good, computing transition states, reaction rates, and exploring free energy barriers with enhanced sampling were relatively poorly supported before this eCSE was completed, despite their importance for a wide range of chemistry and materials science applications. We have implemented two key new features in the CASTEP code to address these issues: (i) support for the widely used nudged elastic band (NEB) transition state search tool, augmented by a state-of-the-art robust optimizer; (ii) an interface to the i-PI universal force engine, which allows CASTEP to be connected efficiently to a range of external codes with enhanced sampling capabilities. The new tools will aid in reconciling experimental observations with atomic-scale behaviour, helping to guide and interpret future experiments. Read more...

Ludwig on ARCHER2: task-based execution
ARCHER2-eCSE01-26 : Prof Davide Marenduzzo (University of Edinburgh)Subject Area:
Chemistry and Materials Computer simulation of fluids - which encompasses both gases and liquids - is important in many contexts. Such computations are the basis of weather and climate forecasts, and many engineering applications such as aerodynamics. At smaller scales, the same ideas can be used to study e.g., blood flow in the body, or to model the behaviour of bacteria and viruses in fluid surroundings. A long-standing collaboration between the Soft Matter Physics Group at The University of Edinburgh and EPCC has developed the Ludwig code for modelling complex fluids. Ludwig can represent a significant range of complexity including solid-fluid interactions, different fluid compositions, and any electric charge carried by the fluid. It can also represent active objects such as simple bacteria. This eCSE project aimed to recast elements of Ludwig to make the code more flexible and therefore able to fully exploit the capabilities of supercomputers. This will allow larger and more complex science cases to be addressed with improved efficiency on ARCHER2. Read more...

Improving the performance of DL_MONTE for large-scale simulations
ARCHER2-eCSE01-19 : Prof Steve Parker (University of Bath)Subject Area:
Chemistry and Materials Monte Carlo molecular simulation (MCMS) entails using random numbers to calculate properties of solids or fluids at the atomic scale. It is the method of choice for studying various physical phenomena of key relevance to technology: common applications include quantifying the amount of a gas which adsorbs to a surface or material; and calculating the atomic-scale properties of fluids and mixtures. There is an insatiable demand for computer simulation to be able to model more complex systems, e.g. systems comprised of more atoms, or more complex molecules. Motivated by this, this project has enabled an open-source MCMS computer program, DL_MONTE, to simulate significantly more complex systems than it could previously. Read more...
Achieving the sustainability and scalability of numeric-atomic-orbital-based linear response and electron-phonon functionality in FHI-aims
ARCHER2-eCSE01-16 : Dr Reinhard Maurer (University of Warwick)Subject Area:
Chemistry and Materials Most calculations run on ARCHER2 will employ electronic structure software packages, which are designed to solve the Schrödinger equation for molecules and materials to obtain their ground state properties. FHI-aims is one such software package, designed to be efficient when running everything from small calculations on standard laptops to huge calculations involving millions of atoms on the largest High Performance Computing systems such as ARCHER2. It is of wide interest to also calculate how molecules and materials respond to atomic displacements, or to electric and magnetic fields. This is possible using density functional perturbation theory (DFPT). A portion of the existing DFPT infrastructure within FHI-aims has been overhauled within this project, to make it more modular, intuitive, scalable, and user and developer friendly. These changes will help researchers to discover and design the next generation of materials more quickly, more cheaply, and more efficiently. Read more...
 and the inhibitor species Y*")
Zacros Software Package Development: Towards Petascale Kinetic Monte Carlo Simulations with the Time-Warp Algorithm
ARCHER2-eCSE01-13 : Dr Michail Stamatakis (University College London)Subject Area:
Chemistry and Materials The chemical industry underpins virtually all sectors of the economy, from healthcare to construction. Catalytic materials, which accelerate reactions, are essential to this industry. Yet discovering catalytic materials and building catalytic processes is non-trivial. Theory and simulation can guide such efforts by delivering fundamental understanding of catalytic function. Zacros is a computational code that simulates chemical events on catalytic surfaces and enables scientists to understand how catalysts accelerate chemical reactions. This eCSE project investigated how the performance of the code is affected by different user-tuneable parameters that affect the efficiency of the parallel algorithm implemented in Zacros. The most important parameter affecting the efficiency of a run was thus identified, and tools were developed that allow users to optimise it for the systems they would be interested in simulating. The improvements made to the code will facilitate further development of improved algorithms and new features in the future. Read more...

CASTEP Solvation Forces
ARCHER2-eCSE01-09 : Prof Matt Probert (University of York)Subject Area:
Chemistry and Materials Density-functional theory (DFT) is a quantum mechanical modelling method which is widely used in physics, chemistry and materials science to study the properties of materials where electrons dictate their behaviour. CASTEP is a UK flagship code which uses DFT to simulate a wide range of properties of materials, such as structure at the atomic level or the vibrational properties of the material. It is one of the most used codes on ARCHER2. Prior to this eCSE project, CASTEP was well adapted to investigating electronic and atomic configurations in solid materials, but not so well suited to investigating isolated molecules, or molecules in solvent, for which there is a wide range of chemistry and materials science applications. The aim of this eCSE project was to add new functionality to CASTEP to address this shortcoming, building on the work carried out in a previous ARCHER eCSE project. The result enables CASTEP users to accurately simulate molecules in solvent, without the cost of explicit solvent molecules. This will therefore open wider avenues of research with CASTEP, for instance, the study of pharmaceutical compounds and future battery designs. Read more...