
Relativistic all-electron orbital-constrained Density Functional Theory to simulate x-ray photoemission and absorption spectroscopy
ARCHER2-eCSE04-03
PI: Prof Reinhard Maurer (University of Warwick)
Co-I(s): Dr Benedikt P. Klein (University of Warwick), Dr Andrew Logsdail (Cardiff University), Dr Volker Blum (Duke University), Dr. Christian Carbogno (Fritz-Haber-Institut der Max-Planck-Gesellschaft), Dr Johannes Lischner (Imperial College London), J. Matthias Kahk (University of Tartu)
Technical staff: Dr Samuel Hall (University of Warwick), Dylan Morgan (University of Warwick)
Subject Area:

Highly scalable first-principles core-level spectroscopy simulations
Core-hole simulations of x-ray photoelectron (XPS) or x-ray absorption spectra (XAS) are able to support the interpretation of complex experimental spectra with overlapping spectral signatures. It is therefore important to have accurate and reliable methods to calculate such spectra. While there are many different methods to do this, core-level-constrained density functional theory (DFT) calculations are the most common and computationally cost-efficient methods. The electronic structure software package FHI-aims has the functionality to perform core-hole calculations and the unique ability to provide absolute binding energy predictions due to its all-electron numeric atomic-orbital basis approach. However, the performance of the calculations suffers from both reliability and memory issues. This is due to neglected code that has not been routinely updated.
The target of this project is to completely overhaul the core-level simulation functionality and to bring it up to date with the current code framework. This has been achieved by migrating all relevant code that was strewn across several modules into one single module. The non-Aufbau Kohn-Sham orbital occupations were determined by outdated routines which caused calculations to be very memory intensive and slow due to lack of parallelisation. Code was repeated several times for different scenarios. In the redesigned code, these occupations are handled by the ELSI library, which allows for fast and memory-efficient calculations. New code has been written in the ELSI code to allow for non-Aufbau occupations needed for a core-hole calculation. All code was collected in a single easily maintained module and almost 3000 lines of redundant code were able to be deleted from the FHI-aims source code. The functionality was further enabled to be used with parallel matrix algebra, which substantially improved scalability to larger systems and on thousands of compute cores on ARCHER2.
This project will benefit the chemistry community by offering an effective way to calculate XPS and XAS for large systems such as metal-organic interfaces which were previously computationally restrictive due to poor scalability. This updated code will make it much easier for new functionality to be implemented in the code relating to core-hole simulations, with one future plan to account for relative effects for higher shell spectra.
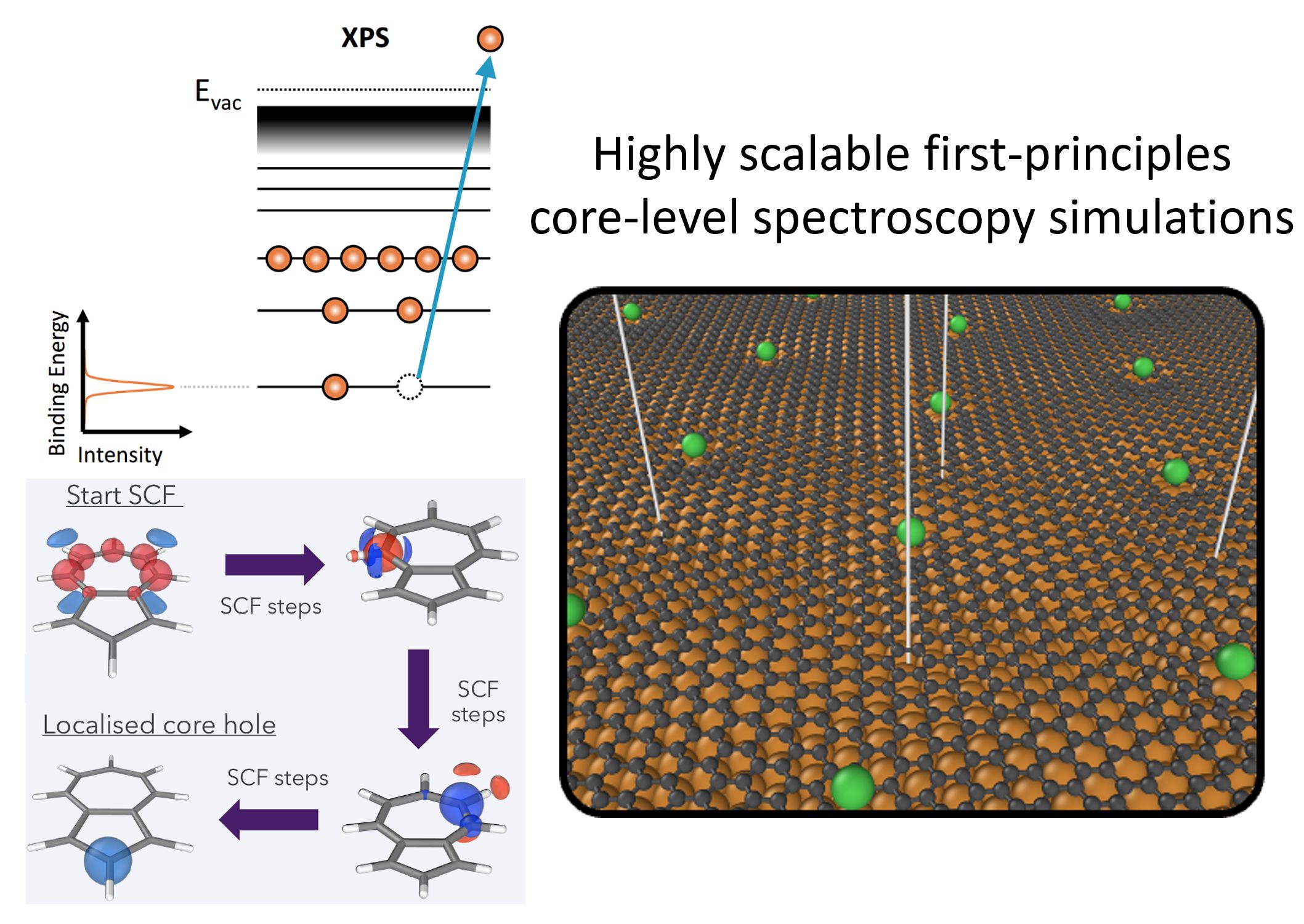
Improvements in parallel high performance computing algorithms enable the simulation of hole formation in core electronic states and the prediction of x-ray photoemission spectroscopy for large scale nanostructured systems.
Information about the code
- The FHI-aims homepage
- Information for obtaining FHI-aims
- The source code for FHI-aims on GitLab
- Specific compilation instructions for ARCHER2
- The tutorial will be accessible on this webpage
Help (both general and functionality specific, including this functionality) is available on the FHI-aims Slack channel, which is available to registered users.