
Computing infra-red spectra using finite differencing in CASTEP
ARCHER2-eCSE09-03
PI: Prof Jonathan R Yates (University of Oxford)
Technical staff: Dr Joseph C A Prentice (University of Oxford)
Subject Area:
Infra-red (IR) spectroscopy is a vitally important tool in understanding the structure of materials relevant to a huge range of industries, from pharmaceuticals and biotechnology to energy materials and earth sciences. It involves shining IR light on the material and seeing how it is absorbed. At certain frequencies, the material is able to absorb the light, at least partially, making the atoms in the material vibrate in a pattern specific to that frequency. The structure of the material significantly affects this process. By experimentally mapping out how strongly the light is absorbed for different frequencies, we can build up an IR spectrum which is characteristic of the material.
However, it can be difficult to work backwards from the experimental spectrum to the structure. To help with this, we can use first principles materials modelling methods – given a proposed structure, we can simulate what the IR spectrum would look like using computational methods. We can then compare this against experiment, and extract further information, such as exactly which atoms are vibrating when a particular frequency is absorbed. Performing such a comparison between computation and experiment is now almost a requirement when using IR spectroscopy in research.
The most commonly used first principles method for simulating the electronic structure of materials, which in turn allows the simulation of IR spectra, is density functional theory (DFT), due to its exceptional balance of accuracy and computational efficiency. There are many well-established codes which implement DFT and DFT-derived methods. This eCSE is focused on CASTEP, a flagship UK-based code which is used extremely widely across both academia and industry. Multiple electronic structure methods are implemented in CASTEP, with the accuracy of the method inversely related to its computational cost and complexity.
A method to use electronic structure calculations to simulate IR spectra, known as density functional perturbation theory (DFPT), was already implemented in CASTEP before this eCSE. DFPT is the preferred method to perform such calculations, but suffers from a significant drawback – a significant amount of mathematical work and coding must be done for each individual electronic structure method. This is not an issue for relatively simple methods, but for advanced methods this practically prevents DFPT from being used.
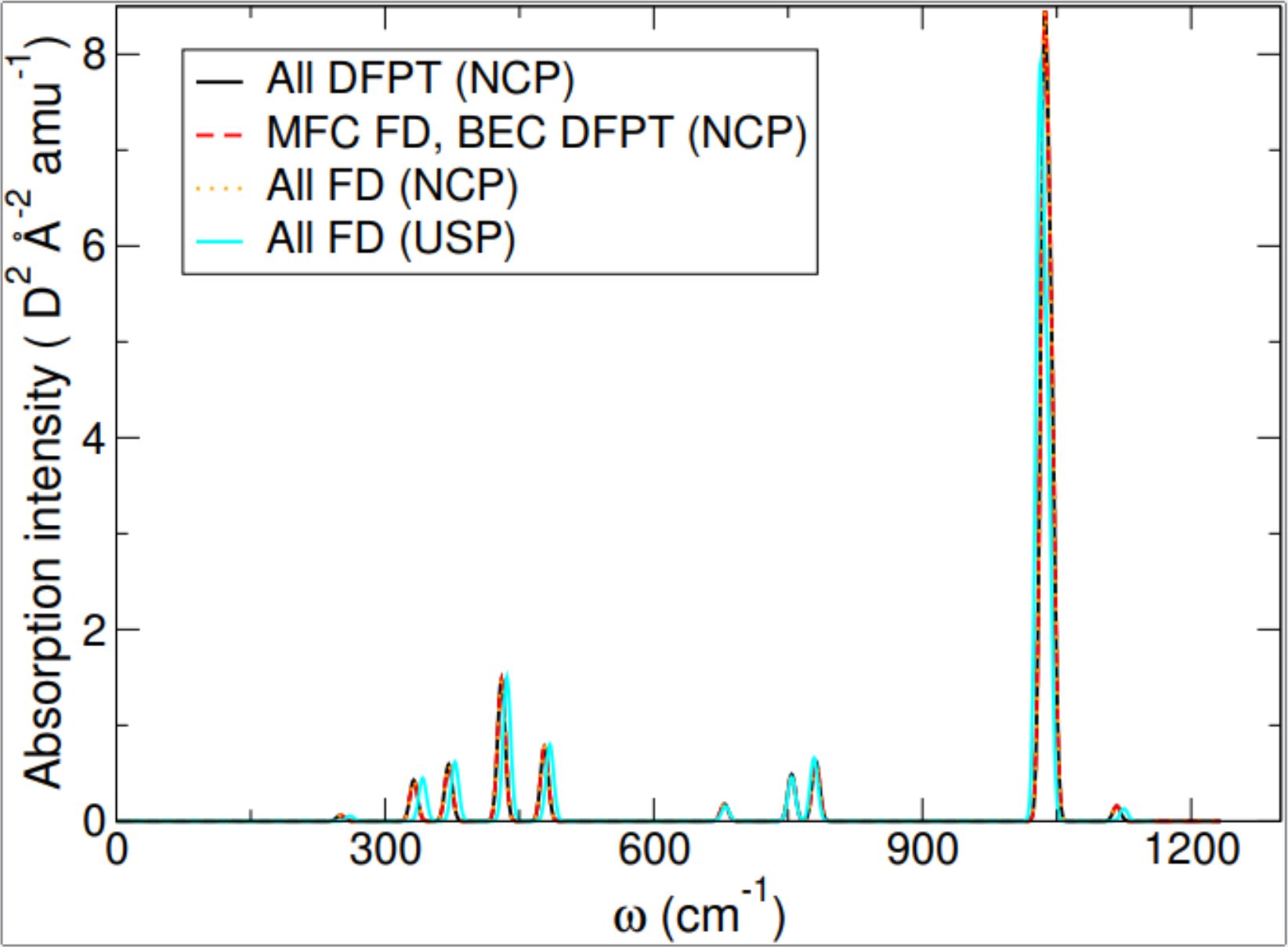
An alternative is to replace DFPT with finite differencing (FD), which has the advantage that it is in principle immediately applicable to any electronic structure method. Whilst the FD approach was already implemented elsewhere in CASTEP, it was not implemented for simulating IR spectra, as such FD IR spectra simulations require the additional step of calculating the electric polarisation of the material. The existing implementation for computing electric polarisation was not compatible with one particular subset of electronic structure methods; it was also not compatible with all the ways CASTEP typically splits up calculations, so that these parts can be run in parallel to speed up the calculation.
This eCSE therefore focused on three main goals: 1. Deriving and implementing the new terms necessary to make polarisation calculations compatible with the remaining electronic structure methods; 2. Making the polarisation calculations compatible with all methods of parallelisation; 3. Implementing the FD method for IR spectra simulations using the modified polarisation code, and making it compatible with all methods of parallelisation. We successfully achieved all these goals, with the additional success of extending our framework to simulate piezoelectricity, implementing them all within CASTEP. This new functionality allows IR spectra to be simulated for materials where advanced methods are a necessity to get even a qualitatively correct result, which would previously have been inaccessible to IR simulation methods. This will be of significant immediate utility to any researchers in both academia and industry using IR spectroscopy, as it opens up new opportunities to compare computational spectra with experimental ones.
Information about the code
Instructions on how to access CASTEP on ARCHER2 can be found on the ARCHER2 website or the CASTEP website