
Zacros Software Package Development: Towards Petascale Kinetic Monte Carlo Simulations with the Time-Warp Algorithm
ARCHER2-eCSE01-13
PI: Dr Michail Stamatakis (University College London)
Co-I(s): Dr Ilektra Christidi (University College London)
Technical staff: Dr Raz Benson (University College London), Dr Giannis Savva (University College London), Dr Ilektra Christidi (University College London), Dr David Stansby (University College London)
Subject Area:
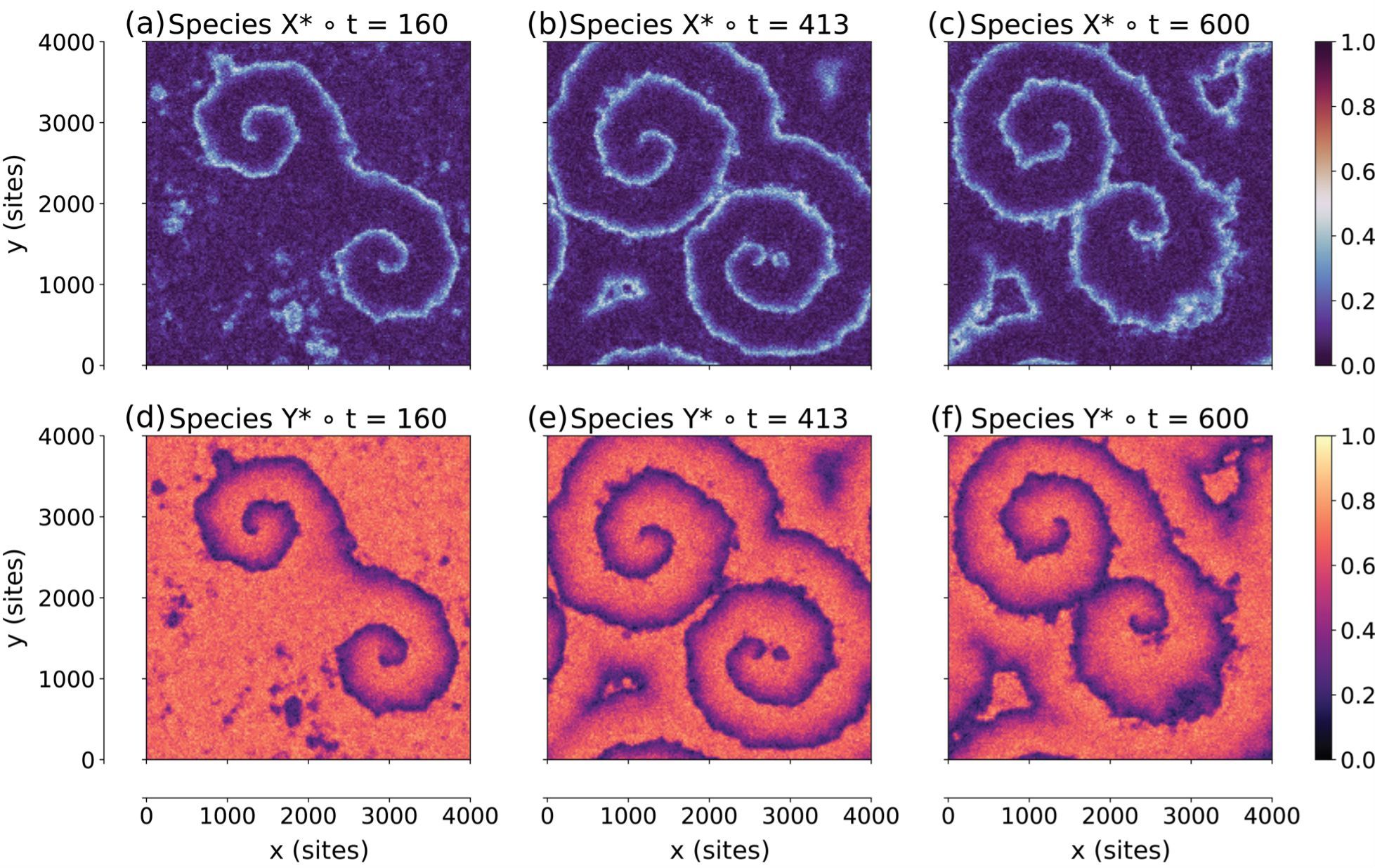
Figure: Snapshots of the fractional coverages of the activator species X* (panels a, b, c) and the inhibitor species Y* (panels d, e, f) at various times (in units of s) during the simulation. Two spirals are reproduced rotating in opposite directions. At time 413 s, a secondary wavefront emerges close to the tip of the lower spiral, and eventually pushes the tip closer to the centre of the domain.
The chemical industry underpins virtually all sectors of the economy, from healthcare to construction, and is an integral part of a successful and sustainable industrial ecosystem. Essential to the chemical industry are catalytic materials, which accelerate reactions and are used by an estimated 85-90% of all chemical manufacturing operations. Yet, discovering catalytic materials and building catalytic processes is non-trivial. Theory and simulation can guide such efforts by delivering fundamental understanding of catalytic function and predictions about the formulation of a catalyst or the operating conditions of a catalytic process that would lead to optimal results.
In this context, Zacros is a computational code that simulates chemical events on catalytic surfaces and enables scientists to understand how catalysts accelerate chemical reactions. It uses the kinetic Monte Carlo (KMC) method, which entails scheduling and executing chemical events in a sequence, at times given by a random number generator. Due to this serial nature of the algorithm, trying to perform such simulations across many processors is challenging. Decomposing a large domain into subdomains and assigning each subdomain to a processor, leads, sooner or later to conflicts in the simulated trajectories of the subdomains, because of events that happen at the shared boundaries of these subdomains. In previous work, we implemented an exact simulation strategy, in the spirit of the so-called “Time Warp algorithm”, which corrects for such conflicts by requiring processors to go back in time and re-simulate “chunks” of the trajectory from the point the conflict arose onwards. To achieve such rollbacks, the processors must periodically keep snapshots of the entire simulation state in the memory, resulting in a high memory footprint for Time Warp simulations. In addition, the user can tune certain operating parameters of the algorithm, such as the frequency of taking snapshots, the memory available for all the snapshots, and the real time interval after which all processors communicate with each other to update the so-called global virtual time, i.e., the time up to which all simulated trajectories are mutually consistent. At these global communication points, processors clean up no-longer-needed snapshots thereby freeing memory.
The current eCSE project focused on understanding how these tuneable parameters affect performance and implemented strategies to lower the memory utilisation and optimise the efficiency and performance of Time-Warp runs. Thus, we found that snapshot saving frequency is the most important parameter affecting the efficiency of a run, and developed tools that allow users to optimise it for the systems they would be interested in simulating. Moreover, we implemented memory amortisation in Zacros, which commits a minimal amount of memory at start-up and increases it on an “as-needed” basis, thereby minimising overall memory utilisation. Finally, we improved the overall quality of our code, which will facilitate further development of improved algorithms and new features in the future.
Publications
- Savva, G. D., Benson, R. L., Christidi, I.-A. and M. Stamatakis (2023). “Large-scale benchmarks of the Time-Warp/Graph-Theoretical Kinetic Monte Carlo approach for distributed on-lattice simulations of catalytic kinetics”. Physical Chemistry Chemical Physics, 25: 5468-5478. (doi: 10.1039/D2CP04424B)
- Savva, G. D., Benson, R. L., Christidi, I.-A. and M. Stamatakis (2023). “Exact Distributed Kinetic Monte Carlo Simulations for On-Lattice Chemical Kinetics: Lessons Learnt from Medium- and Large-Scale Benchmarks”. Philosophical Transactions of the Royal Society A, (doi: 10.1098/rsta.2022.0235)
Information about the code
Any user interested in Zacros can register with XIP, the online licensing portal of UCL Business, access Zacros,, and request a license (this is free-of-charge for academic users). After approval of their request, the user can download the source code and compile it with the Fortran compiler of their choice (GNU Fortran is the preferred option on ARCHER2).