
CASTEP Solvation Forces
ARCHER2-eCSE01-09
PI: Prof Matt Probert (University of York)
Co-I(s): Dr Jacek Dziedzic (University of Southampton), Dr Phil Hasnip (University of York)
Technical staff: Mr Ben Durham (University of York)
Subject Area:
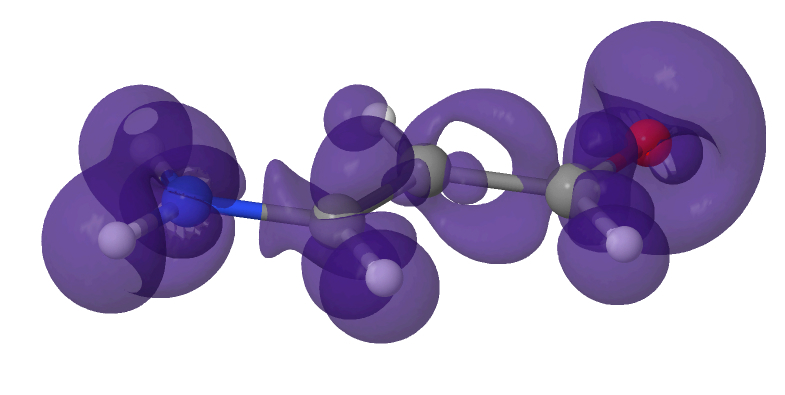
Difference in the electronic density of an organic molecule in a solvated environment compared to in vacuum.
Density Functional Theory (DFT) is a quantum mechanical simulation method that has become one of the most widely used research tools, with approximately 30,000 papers using DFT published each year. The CASTEP DFT code is a UK flagship code, specialised for solid materials, and is heavily used on ARCHER (300-400 active users). CASTEP’s support for obtaining the ground-state electronic and atomic configurations is very good for solid materials with ideal, periodic (repeated) crystal structures. However, computing these properties for isolated molecules or molecules in solvent, for which there is a wide range of chemistry and materials science applications, was poorly supported in the CASTEP code before this eCSE was completed.
In an eCSE project carried out under the previous ARCHER service, the periodic DFT approach of CASTEP was extended to enable simulations in Open Boundary Conditions (OBCs) and in the presence of a solvent, by adopting the minimal-parameter solvent model already developed in ONETEP. However, the OBCs/solvation functionality had some limitations: only the calculation of the electronic ground state was possible, the solvent could not be updated self-consistently with electronic state of the system, and the solving of the OBCs Poisson equation was performed only by the lead MPI process, significantly reducing the parallelism available on the most expensive part of the calculation.
This eCSE project addressed each of these problems. Implementing the calculation of the forces means that it is now possible to optimise the structure in the presence of a solvent, and to perform molecular dynamics simulations to explore the properties and dynamics of the material at finite temperature. This brings the solvation functionality onto much more equal footing with the rest of CASTEP’s functionality. The solvation functionality now has the option to be run such that the solvent is updated self-consistently with the electronic ground-state, although this requires higher sampling and therefore higher memory requirements. The interface to DL_MG (the library used as a Poisson equation solver) was extended to use more of the MPI processes available (in most cases all involved processes), significantly improving the parallel performance and scaling of CASTEP’s OBCs/solvation functionality.
The OBCs/solvation functionality was relatively unknown and under-supported by the CASTEP userbase and CASTEP developer group. Up-to-date documentation has been written to address this, and a tutorial explaining the new functionality has been prepared, along with a webinar which will be uploaded to the CASTEP website after it has been delivered. The increased performance allowed by the extension of the MPI parallelism will also make OBCs/solvation calculations more viable for future usage.
The new code has been merged into the CASTEP development repository and will be included in the forthcoming 2023 release of the code, after which it will be widely available to ARCHER2 users. Key beneficiaries of the project include the CCP-NC, CCP9 and UKCP communities, where the new tools will aid in reconciling experimental observations with atomic-scale behaviour, helping to guide and interpret future experiments.
Information about the code
The new functionality developed in this eCSE project will be included in the upcoming 2023 release of CASTEP.
ARCHER2 users can find information on using CASTEP via the ARCHER2 CASTEP webpage and on the CASTEP website