
HPC Containers?
By Michael Bareford (EPCC) on August 6, 2021
Tags:
Containers are a convenient means of encapsulating complex software environments, but can this convenience be realised for parallel research codes?
Running such codes costs money, which means that code performance is often tuned to specific supercomputer platforms. Therefore, for containers to be useful in the world of HPC, it must be possible to capture this specialisation within a single container. A container should be dedicated to one research code and have the ability to run efficiently on multiple HPC platforms. In this way, containers could facilitate reproducibility and portability by allowing scientists to concentrate on the results of their simulations rather than on the sometimes fiddly compilation scripts necessary for good performance.
Is this a realistic expectation?
Creating a containerized code such that it can run indistinguishably from its non-containerized form is likely to be a painstaking process, sensitive to the choice of code, compiler, MPI library and HPC host. I attempt therefore to answer this question on a case-by-case basis.
GROMACS in a Container on ARCHER2
We start with a containerized GROMACS 2021.1 running on ARCHER2.
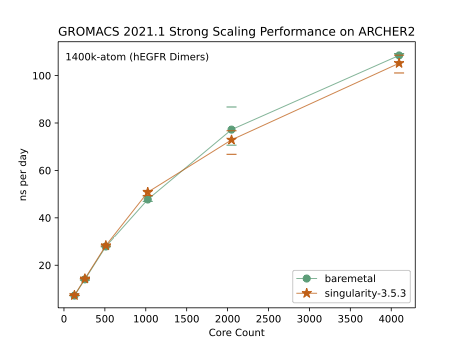
The plot shows the strong-scaling performance for the GROMACS 1400k-atom (hEGFR Dimers) benchmark. This benchmark was run three times for each node count (128 cores per node) and the average plotted - the min and max values are indicated by the horizontal lines. We see that the performance of GROMACS outwith a container, the so-called baremetal configuration, is comparable to the containerised GROMACS. Indeed, on some occasions, GROMACS in a container can beat baremetal. For example, I ran a larger benchmark - 4000k atoms (protein in water) - over 64 nodes (8192 cores) and found that the average performance was 9.47 ns per day for containerized GROMACS and 9.23 for baremetal. These noticeable albeit small differences in performance are surprising given that both codes were built with the same libraries (Cray MPICH v8.0.16 and Cray FFTW v3.3.8.8) using GCC v10 compilers.
One aspect of the benchmark runs that is not under my control is the node assignment, and so, I suspect that for the ARCHER2 4-cabinet system, the differences in compute node performance outweigh any overhead due to containerization.
The following section describes how the GROMACS container was run on ARCHER2.
How to Launch a Containerized GROMACS Job
Part of the Slurm submission script below has been excised for clarity; this is the part responsible for setting up the folder from where the benchmark is run,
see the comments following the module restore command.
#!/bin/bash --login
#SBATCH -J sc_gromacs
#SBATCH -o /dev/null
#SBATCH -e /dev/null
#SBATCH --time=02:00:00
#SBATCH --exclusive
#SBATCH --nodes=16
#SBATCH --tasks-per-node=128
#SBATCH --cpus-per-task=1
#SBATCH --account=<account code>
#SBATCH --partition=standard
#SBATCH --qos=standard
#SBATCH --export=none
# set USER_ROOT, the path off /work pertinent to the user (e.g., /work/z19/z19/mrb4cab/)
# create the folder, APP_RUN_PATH, from where the benchmark will be run
# set APP_OUTPUT, the output file
# set APP_PARAMS, the input parameter string
export OMP_NUM_THREADS=1
# setup singularity and container paths
CONTAINER_PATH=${USER_ROOT}/containers/gromacs/gromacs.sif
# setup singularity bindpaths
APP_SCRIPTS_ROOT=/opt/scripts/app/gromacs/host/archer2
BIND_ARGS=`singularity exec ${CONTAINER_PATH} cat ${APP_SCRIPTS_ROOT}/bindpaths.lst`
BIND_ARGS=${BIND_ARGS},/var/spool/slurmd/mpi_cray_shasta,${USER_ROOT}/tests/gromacs
# setup singularity environment
singularity exec ${CONTAINER_PATH} cat ${APP_SCRIPTS_ROOT}/cmpich8-ofi/gcc10/env.sh > ${APP_RUN_PATH}/env.sh
sed -i -e 's/LD_LIBRARY_PATH/export SINGULARITYENV_LD_LIBRARY_PATH/g' ${APP_RUN_PATH}/env.sh
. ${APP_RUN_PATH}/env.sh
# launch containerised app
RUN_START=$(date +%s.%N)
echo -e "Launching mdrun_mpi (cmpich8-ofi-gcc10) benchmark small (strong) over ${SLURM_NNODES} node(s) from within Singularity container.\n" > ${APP_OUTPUT}
srun --distribution=block:block --hint=nomultithread --chdir=${APP_RUN_PATH} \
singularity exec --bind ${BIND_ARGS} \
${CONTAINER_PATH} /opt/app/gromacs/2021.1/archer2/cmpich8-ofi/gcc10/bin/mdrun_mpi ${APP_PARAMS} &>> ${APP_OUTPUT}
RUN_STOP=$(date +%s.%N)
RUN_TIME=$(echo "${RUN_STOP} - ${RUN_START}" | bc)
echo -e "\nsrun time: ${RUN_TIME}" >> ${APP_OUTPUT}
# cleanup files
...
As far as the srun command is concerned, running a containerized code is not much different to running an executable directly on the compute node.
There are however a couple of preparatory steps. The first of these obtains the host-specific bindpaths held within the container. In the case of
ARCHER2, the bindpaths.lst file contains the following paths.
/work/y07/shared,/opt/cray,/usr/lib64:/usr/lib64/host,/etc/libibverbs.d
The second step gathers the environment settings, again, held within the container; these are specific to particular combination of HPC host,
MPI library and compiler. The environment file env.sh sets the LD_LIBRARY_PATH.
MPI_ROOT=/opt/cray/pe/mpich/8.0.16/ofi/gnu/9.1
...
FFTW_ROOT=/opt/cray/pe/fftw/3.3.8.8/x86_rome
LIBSCI_ROOT=/opt/cray/pe/libsci/20.10.1.2/GNU/9.1/x86_64
BLAS_LIBRARIES=${LIBSCI_ROOT}/lib/libsci_gnu_82_mpi_mp.so
LAPACK_LIBRARIES=${BLAS_LIBRARIES}
LD_LIBRARY_PATH=${FFTW_ROOT}/lib:${LIBSCI_ROOT}/lib:${MPI_ROOT}/lib
LD_LIBRARY_PATH=${LD_LIBRARY_PATH}:/opt/cray/pe/lib64:/opt/cray/libfabric/1.11.0.0.233/lib64
LD_LIBRARY_PATH=${LD_LIBRARY_PATH}:/usr/lib64/host:/usr/lib64/host/libibverbs
LD_LIBRARY_PATH=${LD_LIBRARY_PATH}:/lib/x86_64-linux-gnu:/.singularity.d/libs
...
Notice that the paths starting /opt/cray are pertinent to the host (ARCHER2); that’s because the containerized GROMACS is run according to the
Singularity Bind model. GROMACS was built within the container but against the Cray
MPICH library on ARCHER2. Hence, the containerized GROMACS is started by calling the MPI launcher (srun) from the host. The paths to the MPI
libraries on the host are bound to the container at runtime. The container itself does not have an MPI library.
The version of Singularity currently installed on the ARCHER2 4-Cabinet system is 3.5.3-1, which means that setting LD_LIBRARY_PATH within
the container from a submission script on the host requires one to export the SINGULARITYENV_LD_LIBRARY_PATH variable before calling srun.
Later versions of Singularity permit this to be done by using the --env-file option of the Singularity exec command.
This model can of course be adapted for other MPI libraries such as OpenMPI (see below) which uses a different parallel job launcher, one that
requires a hostfile argument.
...
scontrol show hostnames > ${APP_RUN_PATH}/hosts
chmod a+r ${APP_RUN_PATH}/hosts
...
...
mpirun -n ${NCORES} -N ${NCORES_PER_NODE} -wdir ${APP_RUN_PATH} --hostfile ${APP_RUN_PATH}/hosts \
singularity exec --bind ${BIND_ARGS} \
${CONTAINER_PATH} /opt/app/gromacs/2021.1/archer2/ompi4-ofi/gcc10/bin/mdrun_mpi ${APP_PARAMS} &>> ${APP_OUTPUT}
...
The Container Factory
The details of how the GROMACS container was built without needing root access to ARCHER2 is the subject of a second blog post. Essentially, the container is created at a dedicated Container Factory, a Ubuntu 20.04 instance running within the Eleanor Research Cloud at the University of Edinburgh. Initially, the container features an OS (also Ubuntu 20.04) and the GROMACS 2021.1 source code. The container is then setup as a writable sandbox on ARCHER2 within which GROMACS is built. Following this, the sandbox is converted back to a container image file and downloaded. The whole “targeting” process is directed from the factory and so can be repeated for other HPC platforms. A final GROMACS container could therefore hold multiple executables each one targeting a different HPC host (and MPI library).